


Endoplasmic Reticulum (ER) Stress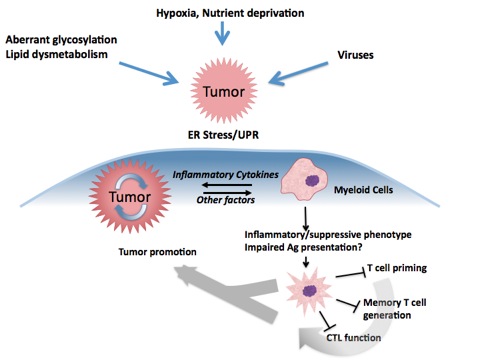
The role of the tumor ER stress response in the regulation of anti-tumor immunity
The UPR is an adaptive cell-signaling pathway that aids eukaryotic cells in coping with conditions in which the protein folding capacity of the ER is saturated or diminished. The tumor microenvironment harbors multiple metabolic ER stressors including low glucose concentration and hypoxia that evoke an ER stress response/unfolded protein response (UPR) in tumor cells, which is critical for tumor cell survival, proliferation, and has been correlated with progression. Based on this evidence, the tumor UPR has been most studied as a cell-intrinsic mechanism by which tumor cells survive.
The laboratory has developed a new paradigm for the initiation of tumor progression, finding evidence suggesting that the tumor cell UPR can function in a cell-extrinsic manner by transmitting ER stress to myeloid cells (macrophages and dendritic cells) that infiltrate the tumor microenvironment, a phenomenon we have termed “transmissible” ER stress (TERS). TERS-stressed myeloid cells upregulate production of tumorigenic, inflammatory cytokines. Furthermore, TERS-primed myeloid cells display a unique functional phenotype, upregulating costimulatory molecule expression, antigen-presentation machinery, and markers of tumor-associated myeloid cells, while downregulating effective high-affinity antigen presentation. Taken together, TERS-primed myeloid cells have many of the characteristics of tumor-infiltrating myeloid cells and thus, we hypothesize that the sequelae of tumor ER stress manifested in infiltrating myeloid cells—i.e. TERS-induced polarization—serve to facilitate tumor growth.
The Zanetti lab has developed two in vitro model systems in which to study transmission of ER stress to myeloid cells and is in the process of validating an in vivo model to study the contribution of TERS to tumorigenesis. The Zanetti lab uses transgenic models of endogenous antigen presentation and cross-presentation coupled with specific anti-MHC/antigen antibodies, and has developed several short-and long-term in vitro T cell priming assays to determine the specific functional consequences of TERS on APCs. With these tools, we hope to elucidate the role of this novel cell-extrinsic phenomenon in dysregulating the function of host immune cells in the tumor microenvironment.
A paper titled “Transmission of ER stress and pro-inflammation from tumor cells to myeloid cells” by Mahadevan, N.R., Rodvold, J., Sepulveda, H., Rossi, S., Drew, A., and Zanetti, M., has been published in Proc. Natl. Acad. Sci. USA and our review on the topic, “Tumor stress inside out: Cell-extrinsic effects of the unfolded protein response in tumor cells modulate the immunological landscape of the tumor microenvironment” by Mahadevan, N.R., and Zanetti, M. is in press in The Journal of Immunology.
Lipocalin 2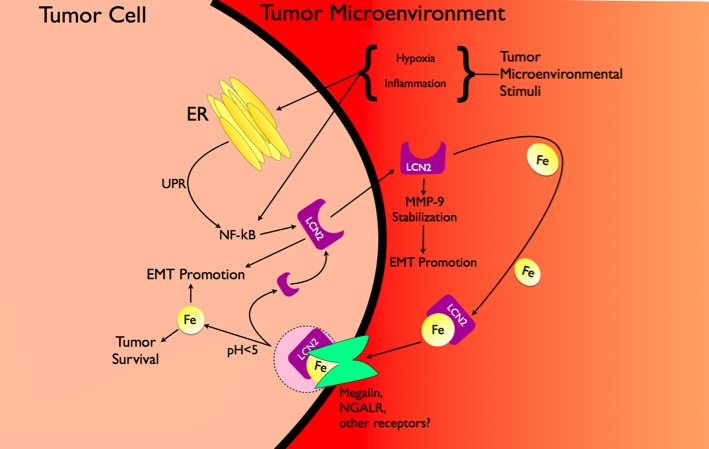
Lipocalin 2 (Lcn2), otherwise known as Neutrophil Gelatinase Associated Ligand (NGAL), was first found as an iron siderophore the immune system secretes in response to bacterial insult. Since its discovery, the small molecule (24 kDa) has become of keen interest in various disease states including diabetes, obesity, bacterial infection, and interestingly cancer. Lcn2 succeeds as an iron siderophore
Despite it's seemingly positive innate immune function, Lcn2 has a malevolent role in tumorigenesis. Its upregulation in various cancers including BCR-ABL leukemia, epithelial tumors of the breast, pancreas, ovary, and lung and gliomas has been correlated to poor clinical prognosis. In breast cancer, Lcn2 is necessary for a tumor’s progression of the epithelial-to-mesenchymal transition (EMT), a hallmark of advanced malignancy. Indeed, it also is a survival factor for thyroid neoplastic carcinoma cells. Despite many reports urging Lcn2 be recognized a potent biomarker for tumor development, very few details about Lcn2’s true functionality in cancer exists.
In a recent microarray, we too found that the Lcn2 gene is heavily upregulated in tumor cells undergoing ER stress, specifically in a mouse prostate carcinoma cell line. In our most recent paper (in review), we report that the unfolded protein response tumor cells innately undergo initiate Lcn2 expression in an NFkB dependent manner. To this end, we propose that a reduction of ER stress would correspond to lower Lcn2 production, thereby dampening its functional, if not elusive, role in tumorigenesis.
The working hypothesis of the lab is that the inherent metabolic stress tumor cells undergo facilitates a significant increase of Lcn2 production and secretion. Once released from the cell, Lcn2 does what it does best: sequesters iron through its siderophore or independently which thereby is then capable of entering the increasing.